MDD - MDR Transition
Until today, medical devices were complying with the Medical Device Directive to sell their products in Europe. However major amendments are made in the current MDD to keep up with the growing technological advances in healthcare and medical devices. The new regulations passed by the Council of the European Union is Medical Device Regulations (MDR) 2017/745 that came into force in May 2017 & manufacturers have a transition time of three years until May 2021 to comply with the new regulations.
IZiel alongwith their partners (Obelis) provide a “One-Stop Shop” to assist medical device manufacturers for MDD-MDR transition. IZiel ensures to focus on regulating medical devices throughout its lifecycle rather than focussing on any one stage, which is the need of MDR. The MDR seeks to address these concerns so that the end users do not face major safety or quality issues.
MDR Timelines
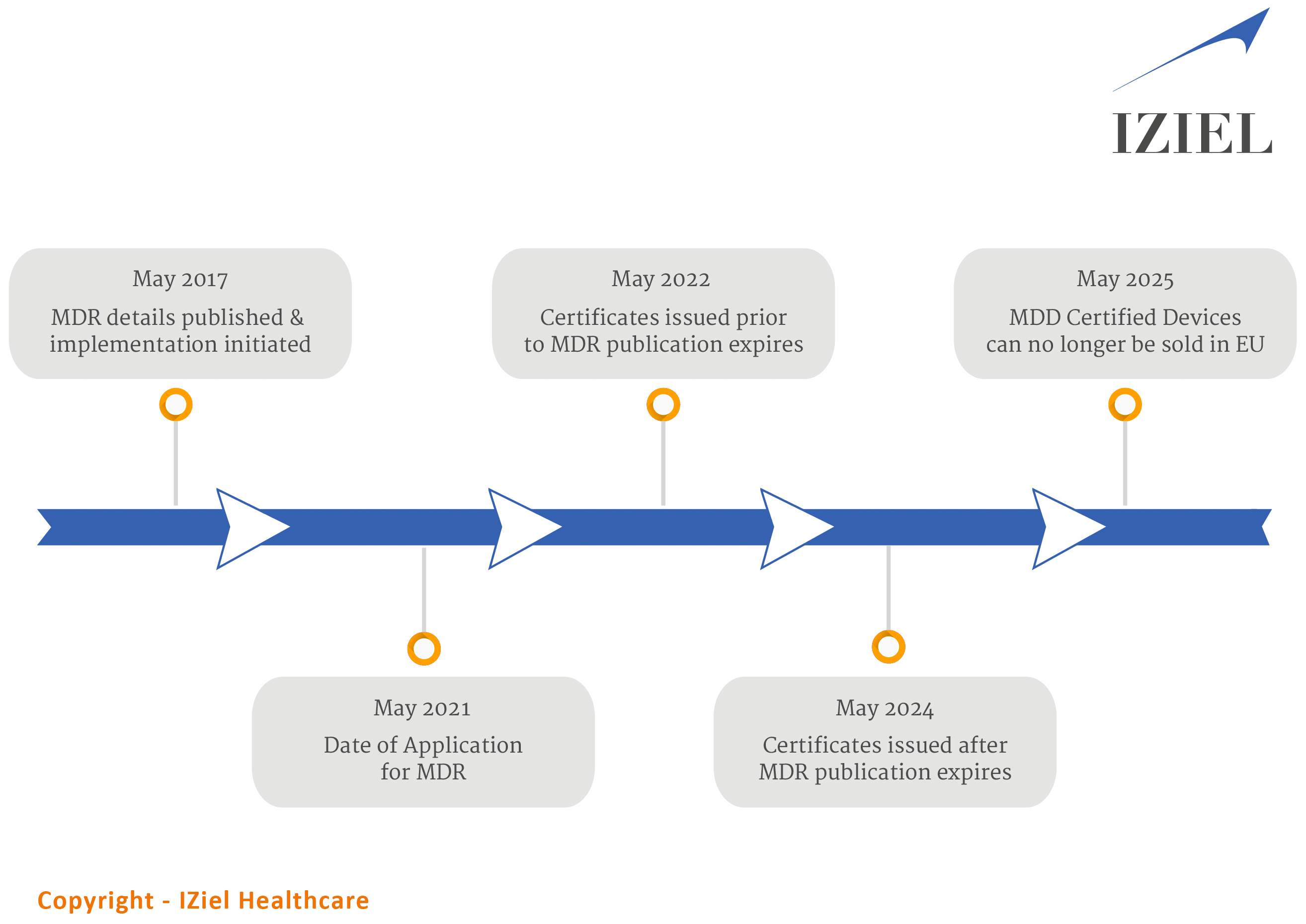 Medical device companies are required to comply with various new / updated regulations of MDR. Many Class I medical device companies are now required to comply alongwith various products are reclassified and all legacy products are required to receive approval too.
Medical device companies are required to comply with various new / updated regulations of MDR. Many Class I medical device companies are now required to comply alongwith various products are reclassified and all legacy products are required to receive approval too.
IZiel Healthcare has collaborated with Belgium based Obelis (European Authorized Representatives) to provide a “One-Stop Solution” to fully support Class I, IIa, IIb & III medical device manufacturers across USA, Europe & Asia. This collaboration would ensure to obtain conformity with the MDR (2017/745) requirements and maintain the CE Marking of global medical devices through technical support, consultancy, representation and device registration services.
Obelis is currently representing over 3000 exporters from more than 60 countries around the world and can provide representation for Europe as well as United Kingdom (UK).
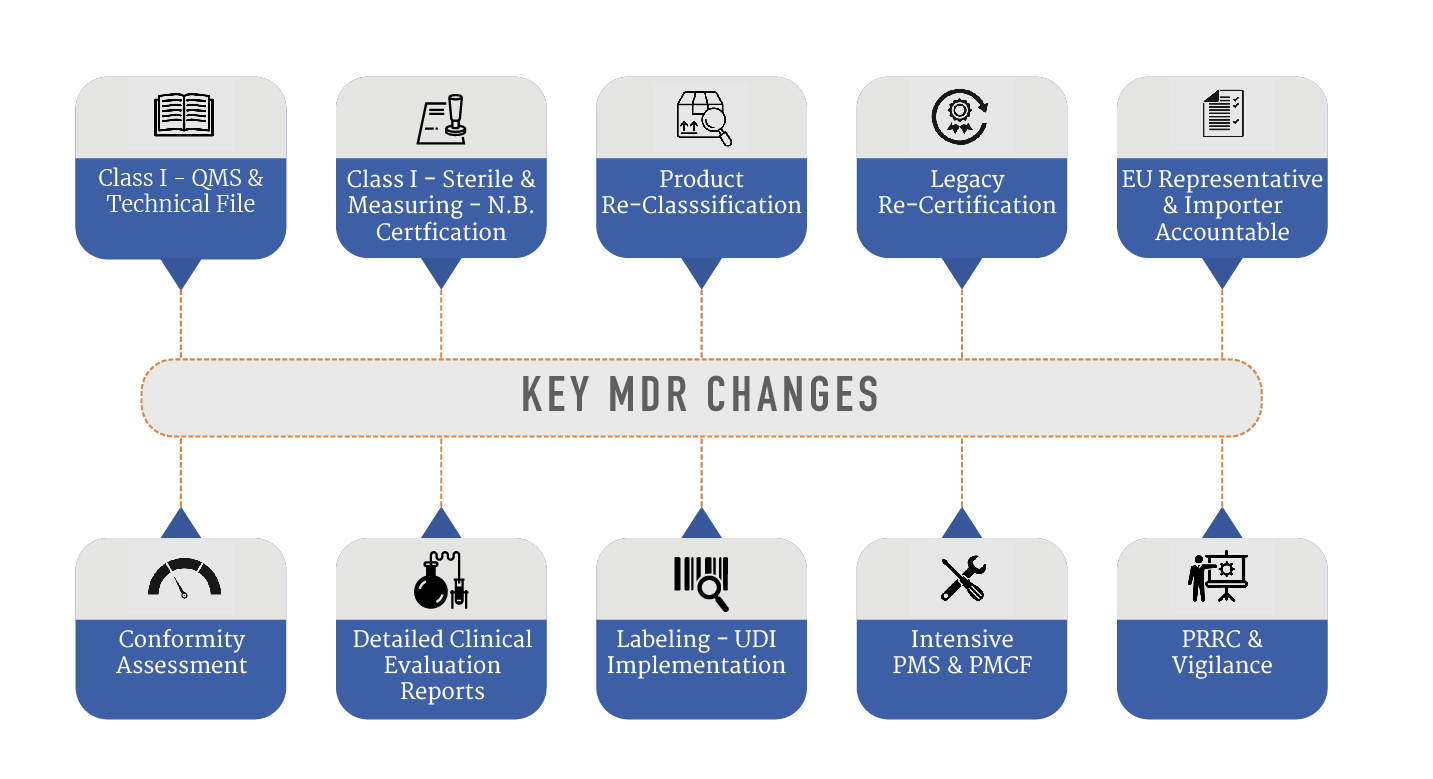
Key MDR Changes include –
 Typically, all MDD-MDR transition projects initiate with Gap Assessment. Gap Assessment is a crucial activity during MDR transition & our team with engineering & regulatory expertise are well equipped to conduct this activity with an analytical mindset, resolve any regulatory concern and develop robust regulatory strategy for medical device manufacturers.
Typically, all MDD-MDR transition projects initiate with Gap Assessment. Gap Assessment is a crucial activity during MDR transition & our team with engineering & regulatory expertise are well equipped to conduct this activity with an analytical mindset, resolve any regulatory concern and develop robust regulatory strategy for medical device manufacturers.
Our “ONE-STOP COMPLETE SOLUTION” include –
- Gap Assessment
- Technical File Preparation
- QMS Documentation
- Expert Review & Recommendations
- European Authorized Representative (EC REP)
- EUDAMED
- Mock Audits & Trainings
- PRRC Services
- Clinical Evaluation Plan & Report
- Software Validation
IZiel team of specialists and quality professionals look forward to support more medical device companies to file their devices under the MDR 2017/745.