USFDA Approvals for Medical Devices
Medical Device Manufacturers require USFDA Approvals to sell their products in USA. USFDA differentiates product approvals in Class I, II &III depending upon the risk associated. The submissions include self-certification, 510(k) and PMA depending upon the class of the product.
IZiel implements an Outcome-Based Delivery Model to provide complete solution from developing engineering documentation to receiving USFDA Approvals & establishment registrations thereafter. Our teams from USA & India collaborate to develop a Cost-Effective Model to complete USFDA Approvals for customers in USA, Europe and Asia.
USFDA Approval Requirements
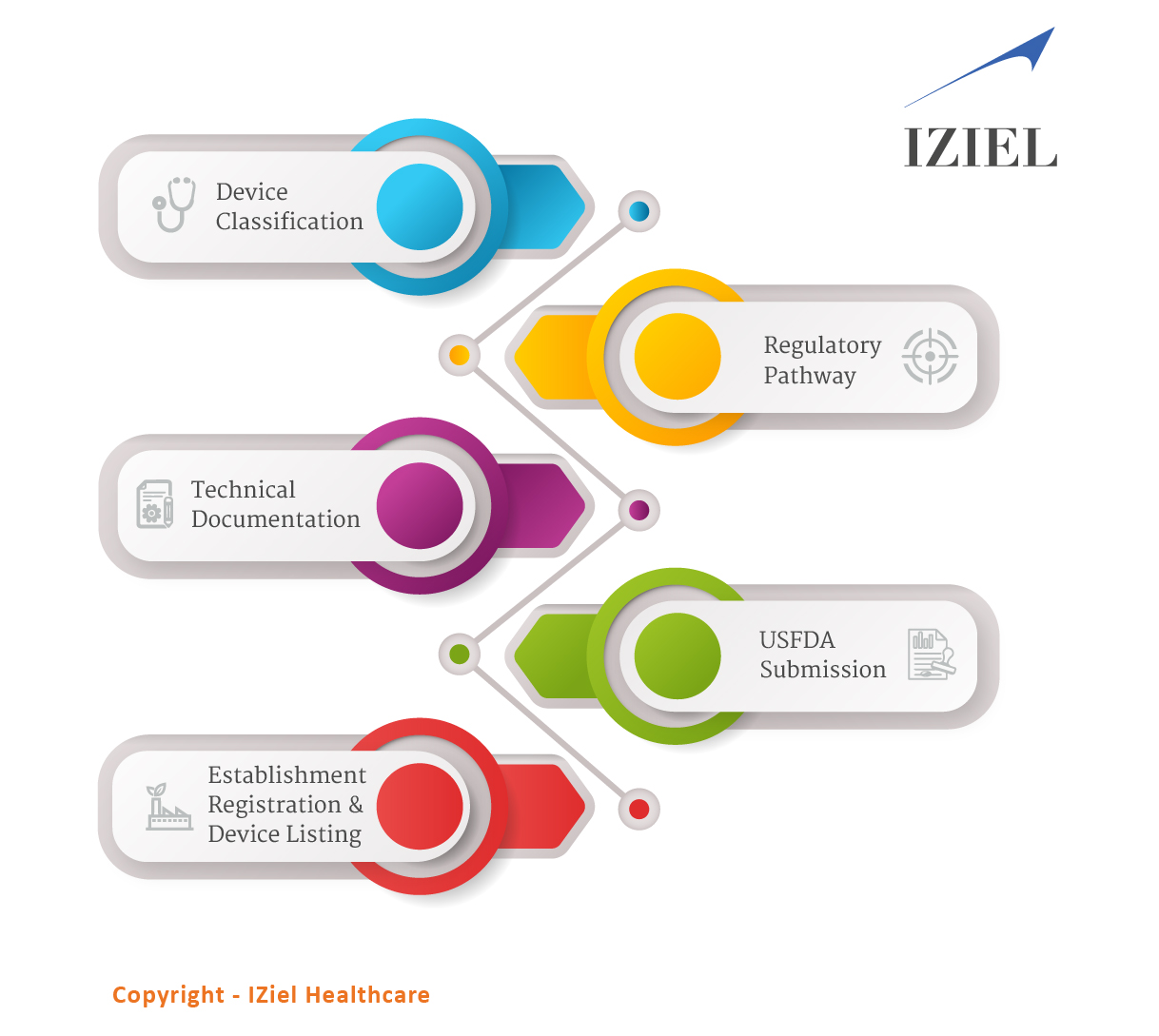
IZiel adopts an analytical mindset thus enabling us to root out all possible non-conformances in a regulatory submission. IZiel works in collaboration with your team to develop the complete Design History File (DHF) including requirements management, risk management, process validations and software validations using robust design controls process and quality system procedures. Thereafter, IZiel team works with their regulatory team in USA to complete the submissions (510k or PMA) for USFDA Approvals.
Following illustration would help you to understand the requirements of USFDA Approvals –
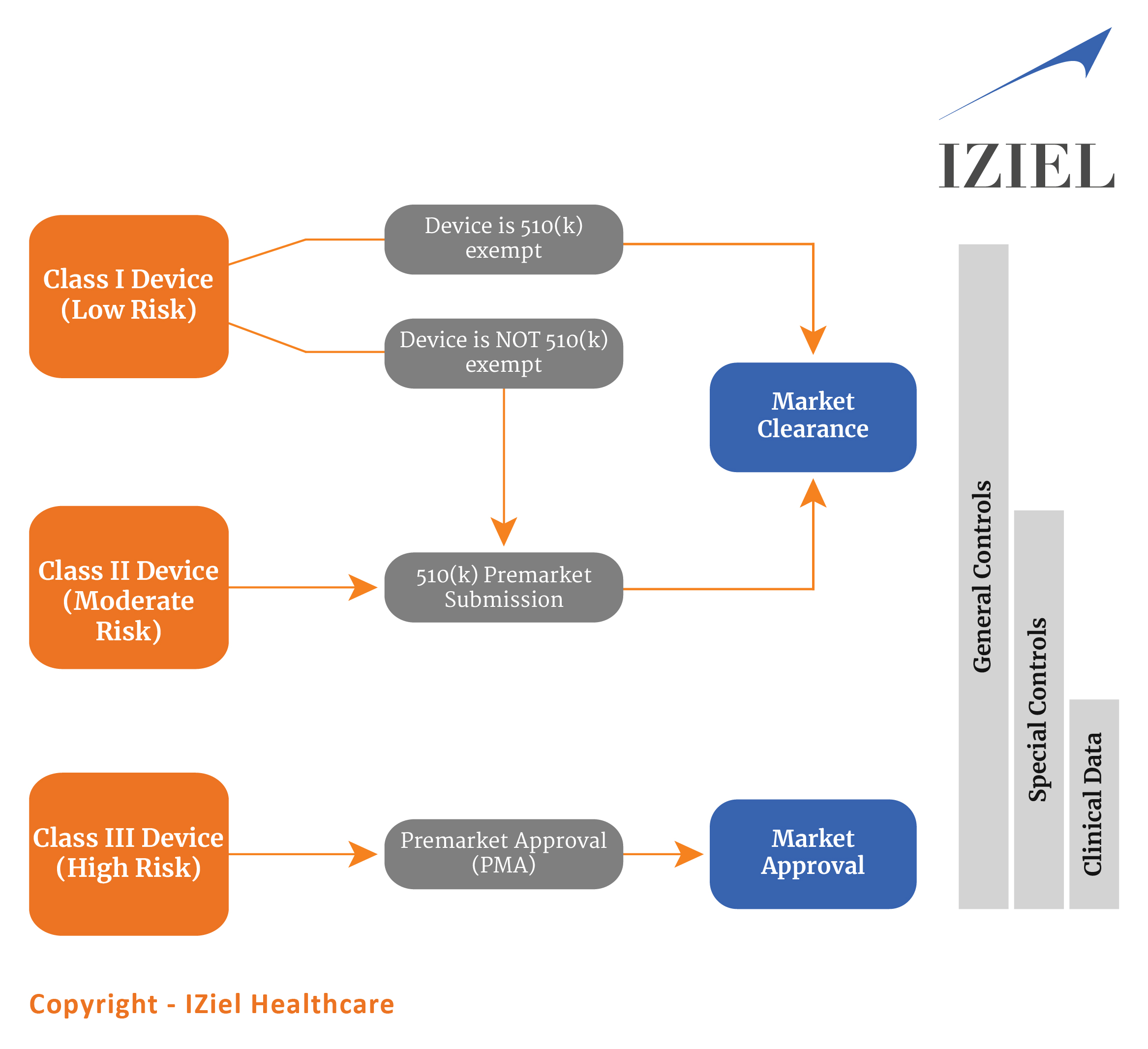
Upon completion of Engineering & QMS Documentation, the regulatory team of IZiel conducts the predicate device search, comparative analysis and ensure all the necessary standards are complied with. Thereafter, IZiel team initiate the writing of 510(k) / PMA, which is verified and approved by our US Regulatory Consultants. Our highly experienced US regulatory consultants include Ex-FDA Auditors, have conducted 3500 + reviews and approvals and have experience with Class I, II & III devices in Cardiology, Neurology, Image Diagnostics and Orthopaedic products.